

A atrofia muscular espinhal (AME) é uma grave doença degenerativa geneticamente determinada que se caracteriza pela perda progressiva dos motoneurônios localizados no corno anterior da medula espinhal e na coluna eferente somática de núcleos de nervos cranianos no tronco encefálico. Essa degeneração dos motoneurônios leva a fraqueza muscular progressiva, atrofia dos músculos esqueléticos e graus variáveis de insuficiência ventilatória. A forma mais comum de AME é causada por mutações no gene de sobrevida do neurônio motor 1 (SMN1), e é herdada em um padrão autossômico recessivo.
Aproximadamente 95 a 98% dos indivíduos com o diagnóstico clínico de AME apresentam deleção do éxon 7 em ambas as cópias do gene SMN1, enquanto cerca de 2 a 5% dos pacientes apresentam deleção do éxon 7 em um dos alelos do gene e uma variante intragênica no outro alelo. O tratamento até então era baseado em fisioterapia e cuidados multidisciplinares com ênfase na parte respiratória e nutricional. No entanto, nos últimos anos várias pesquisas resultaram em descobertas de tratamentos medicamentosos eficazes no controle da progressão da doença e até mesmo na melhora de funções motoras e respiratórias.
Como é o sistema de classificação da AME e quais os principais sintomas destes subtipos?
A AME é classicamente classificada em pelo menos quatro tipos baseando-se na idade de início e na função motora máxima obtida pelo paciente. AME tipo 1, ou doença de Werdnig Hoffmann, é o quadro mais grave com início das manifestações nos primeiros 6 meses de vida e no qual a criança não alcança a habilidade motora de sentar independentemente. Na AME tipo 2 as manifestações iniciam após 6 meses de idade e antes se completar o primeiro ano de vida; as crianças são capazes de sentar, mas não adquirem a habilidade de andar independentemente. AME tipo 3, também conhecida como doença de Kugelberg Welander, tem apresentação clínica iniciada no segundo ano de vida ou após, e os pacientes adquirem a capacidade de andar em algum momento da vida. AME tipo 4 é uma forma mais leve, com fraqueza muscular de início na vida adulta. É também descrita uma forma mais rara e extremamente grave de AME, o tipo 0, com manifestações notadas ainda durante a gestação e sintomas ocorrendo nos primeiros dias de vida.
O que é o gene SMN2 e qual sua diferença para o gene SMN1?
O gene SMN2 é muito parecido com o gene SMN1, mas não é capaz de produzir a mesma quantidade da proteína SMN funcional. Os pacientes com AME tem mutação no gene SMN1, mas retém o gene SMN2. No entanto, a presença do gene SMN2 não é suficiente para evitar a doença, pois a quantidade de proteína funcional produzida por este gene é pequena. Alguns pacientes têm mais cópias do gene SMN2, então produzem uma quantidade maior da proteína SMN e, portanto, apresentam um quadro clínico mais leve. Em geral, os pacientes com AME tipo 1 apresentam duas cópias do gene SMN2, os com AME tipo 2 apresentam usualmente 3 cópias do gene SMN2 e os pacientes com AME tipo 3 possuem de 3 a 4 cópias do gene SMN2. Sabendo disto, os pesquisadores há alguns anos tem tentado “ativar” o gene SMN2 a produzir mais proteína funcional e melhorar o quadro clínico dos pacientes.
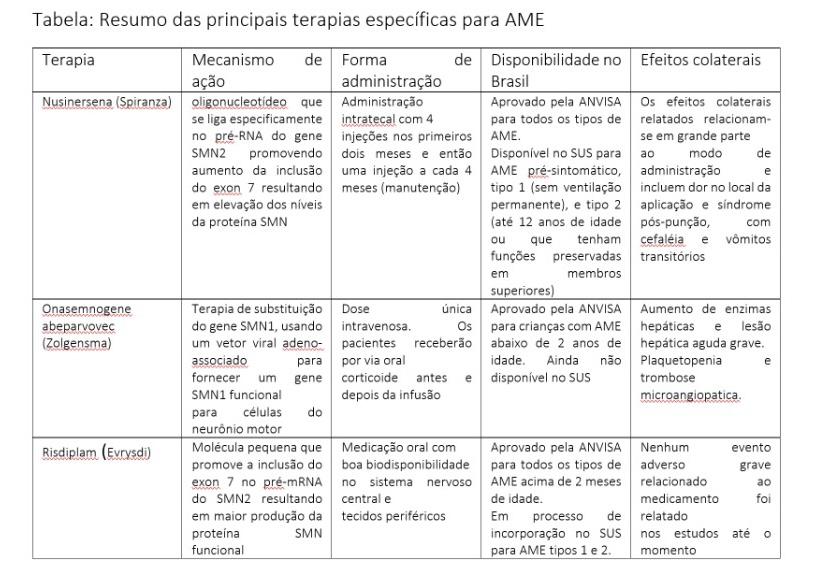
Como agem e quais são os efeitos do Nusinersena e do Risdiplam?
Nusinersena (Spinraza®) é um oligonucleotídeo antisense de administração intratecal aprovado para o tratamento da AME (Tabela). O medicamento modifica o splicing de RNA pré-mensageiro do gene SMN2, aumentando assim os níveis de proteína SMN funcional. Nos estudos de fase 3 (CHERISH e ENDEAR), o Nusinersena alcançou resultados significativos e/ou clinicamente relevantes, produzindo melhora na função motora em pacientes AME tipo 1 e de início tardio, e melhora significativa na sobrevida livre de eventos e na sobrevida global em pacientes com AME tipo 1. A experiência de mundo real tem confirmado a eficácia, segurança e tolerabilidade de Nusinersena em pacientes com AME de todas as idades, e tem demostrado manutenção dos efeitos da terapia a longo prazo. São poucos os efeitos colaterais do Nusinersena, e usualmente relacionados com a aplicação intratecal, tais como, vômitos e dor de cabeça, por algum período após a injeção. Outros efeitos colaterais descritos incluem aumento das infecções respiratórias e alteração na coagulação sanguínea.
O Risdiplam (Evrysdi®) também aumenta a produção de proteína SMN funcional através de modificação no splicing de pré-RNA mensageiro do gene SMN2. No entanto, seu sítio de ligação é diferente do sítio de atuação do Nusinersena. O Risdiplam é um composto capaz de atravessar a barreira hematoencefálica, desta forma, sua administração é por via oral (TABELA). Estudos de fase 2/3 mostraram a eficácia do medicamento no tratamento de crianças com AME de diferentes tipos e idades. No estudo FIREFISH que envolveu 41 lactentes com AME tipo 1, após 12 meses de tratamento, 12 lactentes (29%) conseguiram sentar sem apoio por pelo menos 5 segundos. O uso do Risdiplam resultou em porcentagens mais altas de lactentes que atingiram marcos motores e que apresentaram melhora na função motora do que as porcentagens observadas em coortes históricas de pacientes não tratados, e 85% das crianças estavam vivas e fora de ventilação mecânica permanente após 12 meses de tratamento. O estudo SUNFISH incluiu 120 pacientes com AME tipo 2 ou tipo 3 não ambulantes e idade entre 2-25 anos tratados com Risdiplam em comparação com 60 pacientes controles. Risdiplam resultou em uma melhora significativa na função motora nos pacientes tratados. A função motora foi geralmente melhorada em indivíduos mais jovens e estabilizada em indivíduos mais velhos. Nestes estudos, o Risdiplam foi geralmente bem tolerado, com um benefício favorável em relação ao risco.
As pesquisas clínicas com Nusinersena e Risdiplam mostraram que quanto mais precoce o tratamento é iniciado, melhor é o efeito. As crianças com AME tipo 1 que receberam as terapias passaram a adquirir funções motoras que não teriam se não tivessem recebido o medicamento, por exemplo, sustentar a cabeça, sentar e até mesmo andar. Quando as terapias foram administradas antes que a doença comece a se manifestar, em uma fase pré-sintomática, estudos tanto com o Nusinersena (estudo NURTURE) quanto com o Risdiplam (estudo RAINBOWFISH), tem mostrado resultado excelentes, com a maioria das crianças evoluindo com mínimos sinais clínicos da doença; próximo da normalidade.
Como age e quais são os efeitos da terapia gênica?
A terapia gênica é um tratamento que melhora os níveis de proteína SMN funcional através da introdução na célula do paciente de um novo gene SMN1 (TABELA). Nesta terapia, uma cópia do gene SMN1 é inserida em um vetor viral AAV9 não replicante. O vetor AAV9 é capaz de atravessar a barreira hematoencefálica, permitindo assim a administração endovenosa. Como o fragmento de DNA inserido fica retido no núcleo da célula, o medicamento é aplicado em uma só dose. No entanto, a duração do efeito da terapia ainda não foi estabelecida.
Estudos pivotais demonstraram eficácia do tratamento com Onasemnogene abeparvovec (Zolgensma®) para crianças com AME tipo 1. O STR1VE-USA foi um estudo aberto de fase 3, de braço único e dose única, realizado em 12 hospitais e universidades nos EUA; e confirmou resultados de eficácia do estudo START de fase 1. Vinte e dois pacientes foram elegíveis e receberam Onasemnogene abeparvovec. Treze pacientes (59%) alcançaram a habilidade de sentar independente por 30 segundos ou mais na visita do estudo de 18 meses de idade. Vinte pacientes (91%) sobreviveram sem ventilação permanente aos 14 meses. No estudo STR1VE-EU realizado em centros europeus, dentre 32 pacientes incluídos, 44% atingiram a capacidade de sentar por 10 segundos aos 18 meses de vida, e 97% deles se mantiveram livres de ventilação mecânica aos 14 meses de idade. Nos dois estudos, o evento adverso mais comum foi infecção de vias respiratórias, enquanto o evento adverso mais comum relacionado com a medicação foi elevação de transaminases séricas. Dados de vida real publicados mais recentemente tem confirmado a eficácia da medicação em manter, ou mesmo melhorar, funções motoras, bulbares e respiratórias de crianças com AME até 24 meses de idade. No entanto, com o aumento da idade das crianças incluídas nos estudos, eventos adversos tem sido mais frequentes e mais graves. Em especial, a elevação das enzimas hepáticas tendem a aumentar significativamente com a idade e o peso da criança. Microangiopatia trombótica é outra complicação potencial grave relatada em crianças que usaram o medicamento.
Para serem elegíveis para receber Onasemnogene abeparvovec, os pacientes com AME devem ser testados quanto à presença de anticorpos anti-AAV9 pré-existentes, que podem, em teoria, aumentar o risco de respostas imunes clinicamente relevantes. Os pacientes com AME devem ter títulos de anticorpos anti-AAV9 não superiores a 1:50, conforme medido por ensaio imunoenzimático.
Ensaios clínicos publicados e dados de mundo real para a administração do Onasemnogene abeparvovec tem se concentrado na monoterapia em populações homogêneas e restritas, limitando a generalização dos resultados do tratamento. A dose do vetor de terapia gênica é dependente do peso, assim a segurança e eficácia do Onasemnogene abeparvovec em uma população mais heterogênea de crianças mais velhas, mais pesadas e/ou sintomáticas recebendo terapias combinadas ou sequenciais ainda não está bem definida. Desta forma, quando administrado após os 6 meses de idade e/ou em estágios avançados da doença, os pais e médicos devem estar cientes de que até o momento não há dados publicados sobre eficácia e segurança. Nesta população de pacientes, é particularmente importante que os médicos discutam a relação benefício/risco e gerenciem cuidadosamente as expectativas dos pais ou pacientes.
Há evidências de ganho adicional na eficácia clínica com a utilização combinada destas terapias?
Não há evidências científicas até o momento que mostrem vantagens na utilização de terapias combinadas para tratamento da AME. Há na literatura médica relatos de pequenas coortes de pacientes em uso de terapias combinadas, com resultados conflitantes, no entanto, tais estudos possuem pouco tempo de uso destas terapias além de número pequeno de indivíduos testados.
As terapias atuam como cura para AME?
Nenhuma destas terapias funciona como cura definitiva da doença. O Nusinersena e o Risdiplam atuam modulando o splicing do RNA mensageiro do gene SMN2. Assim, ao suspender o uso destes medicamentos, o efeito benéfico é eliminado. A terapia gênica atua adicionando no núcleo das células do neurônio motor, uma sequência de DNA do gene SMN1. Tal sequencia de DNA fica livre no núcleo na forma de um episomo, e portanto não é incorporado ao DNA do paciente. O tempo do efeito desta terapia não é determinado, e a taxa de eliminação deste episomo do núcleo das células dos pacientes também não é conhecida. Desta forma, não é possível afirmar que a terapia gênica cura definitivamente o indivíduo com a doença.
Estas terapias poderiam ser usadas “off label” para tratamento de outras doenças musculares degenerativa ou são específicas para AME?
Estas medicações são específicas para AME. No caso da terapia gênica, há inserção de um gene SMN1 homólogo no DNA do paciente. Enquanto tanto o Nusinersena quanto o Risdiplam atuam no RNA mensageiro modulando especificamente o gene SMN2.
Considerando o alto custo das terapias, seria uma estratégia razoável criar critérios de priorização para prescrição destas terapias?
Este é um assunto complexo e controverso. Embora, a eficácia das terapias não tenha sido adequadamente avaliada para alguns grupos de pacientes, tais como aqueles com longo tempo de doença, traqueostomizados, em ventilação permanente, e com importantes deformidades osteoesqueléticas, até o momento não há evidências científicas indicando que estes mesmos grupos de pacientes definitivamente não se beneficiariam do tratamento. A impressão que todos os estudos têm demonstrado, é que pelo menos há uma estabilização na progressão da doença, e isto por si só já seria uma indicação para a sua prescrição. Além do mais, para cada paciente há prioridades próprias. Por exemplo, para alguns a prioridade pode ser melhorar a fala, a deglutição e a capacidade de tocar a cadeira de rodas, e não só apenas andar, sentar ou ficar fora da ventilação mecânica. Desta forma, no momento não é possível determinar critérios de priorização para prescrição de terapias específicas para AME.
Qual a importância do diagnóstico genético?
Como comentado anteriormente, quanto mais precoce o início do tratamento, melhor é o efeito do medicamento. Assim, o diagnóstico genético deve ser feito o mais precocemente possível. Em alguns países já tem sido realizado inclusive triagem neonatal na tentativa de identificar indivíduos em fase pré-sintomática. O teste genético, além de determinar se o paciente tem a mutação no gene SMN1, também vai indicar o número de cópias do gene SMN2, e esta informação é importante para dar alguma sugestão do prognóstico.
Porque este tema também é relevante para o neurologista?
A AME é uma doença predominantemente infantil. No entanto, com um tratamento de suporte adequado, estas crianças chegam facilmente a idade adulta, em especial aquelas com os tipos 2 e 3. Por ser uma doença com manifestações predominantemente motoras, não é infrequente que o primeiro Médico a ser solicitada avaliação seja o Neurologista. Desta forma, ter conhecimento sobre a doença torna-se importante no sentido de promover um diagnóstico mais precoce e assim iniciando tratamento adequado.
* Edmar Zanoteli – Professor do Departamento de Neurologia da Faculdade de Medicina da Universidade de São Paulo.
Este texto foi baseado na experiência do autor e em artigos científicos publicados na literatura Médica: Paik J. Risdiplam: A Review in Spinal Muscular Atrophy. CNS Drugs. 2022 Mar 13. doi: 10.1007/s40263-022-00910-8. Pitarch Castellano I, et al. Delphi consensus on recommendations for the treatment of spinal muscular atrophy in Spain (RET-AME consensus). Neurologia (Engl Ed). 2022 Feb 28:S2173-5808(22)00012-8. Abbas KS, et al. The Safety and Efficacy of Nusinersen in the Treatment of Spinal Muscular Atrophy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Medicina (Kaunas). 2022 Feb 1;58(2):213. Aslesh T, Yokota T. Restoring SMN Expression: An Overview of the Therapeutic Developments for the Treatment of Spinal Muscular Atrophy. Cells. 2022 Jan 26;11(3):417. Flotte TR, Gessler DJ. Gene Therapy for Rare Neurological Disorders. Clin Pharmacol Ther. 2022 Apr;111(4):743-757.









